Introduction
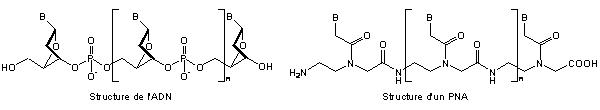
Two compared backbones of DNA (left) and deltapeptidic PNA (right) Peptidonucleic acids (PNA) are DNA mimics with a pseudopeptide backbone. PNA is an extremely good structural mimic of DNA (or RNA), and PNA oligomers are able to form very stable duplex structures with Watson-Crick complementary DNA, RNA (or PNA) oligomers. Usually, PNA are built on a delta peptide backbone, in order to optimize tha spacing between two stacked nucleic bases. In our laboratory, peptidic synthesis is carried on on N-aminopeptides, in order to synthesize peptidonucleic acids based on an N-aminopeptidic backbone.
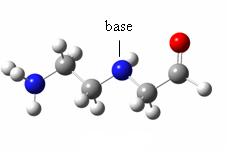 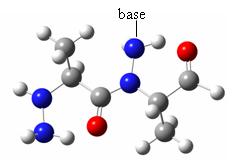
deltapeptidic backbone(left) and N-aminopeptidic backbone (right) In the present study, we want to model the interactions between the N-aminopeptidic backbone and the amide linkages used to serve as arms bearing the nucleic bases, in order to assess the stability of a peptidonucleic acid building block, with regard to its conformational freedom.
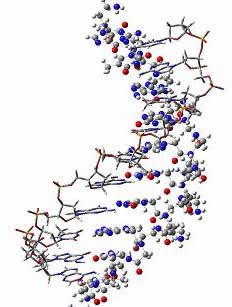
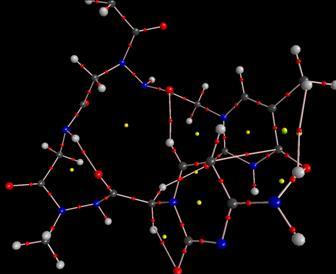
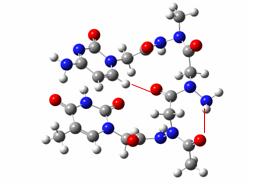
PNA-DNA duplex showing the DNA part in sticks and the PNA part in balls and sticks
To do this, we extracted a PNA-DNA duplex structure from the PDB database, erased the DNA moiety of the structure, changed the deltapeptidic backbone to a N-aminopeptidic one, and then optimized the geometry of the macromolecule at the molecular mechanics level, with every nucleic base frozen to its initial geometry in order to mimic a complex with a DNA moiety.
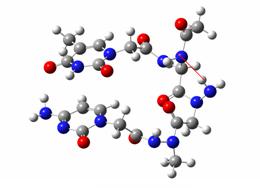
It is important to note that this molecular mechanics optimization is only a preliminary study as the N-N bond is very poorly defined by molecular mechanics.
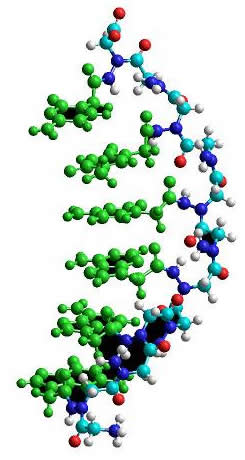
The molecular mechanics optimized N-amino PNA structure showing frozen parts (in green)
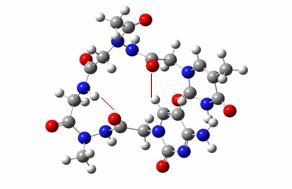
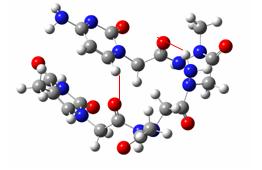
The configurational position (cis or trans) of every peptidic link was then assessed and different assemblies of monomers (the moieties linking one nucleic base to another through two arms linked to the peptidic backbone) were defined according to their cis or trans configuration. Four kinds of building blocks were thus defined, and then optimised at the B3LYP/6-31G(d,p)//HF/STO-3G level of theory and their energies compared. To find reasons for energetical differences, an AIM study of the electronic density of the optimized structures has then been carried on, focussing on the intramolecular hydrogen bonding network.
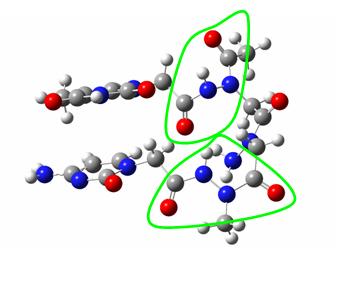
A monomer building block showing the four peptidic linkages in cis or trans configuration
(note that the backbone central peptidic link is always trans)
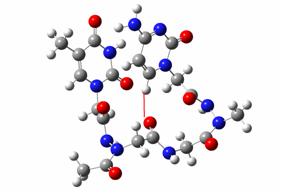
In this case, the peptidic links from upper left (thymine) to down left (cytosine) are
trans then cis then trans then trans. It is thus named :
Thy-trans-cis-trans-trans-Cyt
Admittedly, the present study does not model the role of the solvent around this typical biomolecular system. The idea is to get hints about intrinsic reasons for conformational stability of the studied building blocks, as opposed to external reasons from the solvent or other influences.
|